Cancer / inflammation
PROTAC technology to investigate dysregulation of the Ubiquitin-Proteasome System
Carlotta Cecchini 1, Margaux Héritier 1, Valentina Ceserani 2, Federico Costanzo 2, Jean-Philippe Theurillat 2, Francesca Tessaro 1, Sébastien Tardy 1 and Leonardo Scapozza 1
1 School of Pharmaceutical Sciences, University of Geneva, Rue Michel-Servet 1, 1206 Genève
2 Functional Cancer Genomics Group, Institute of Oncology Research, Via Vela 6, 6500 Bellinzona
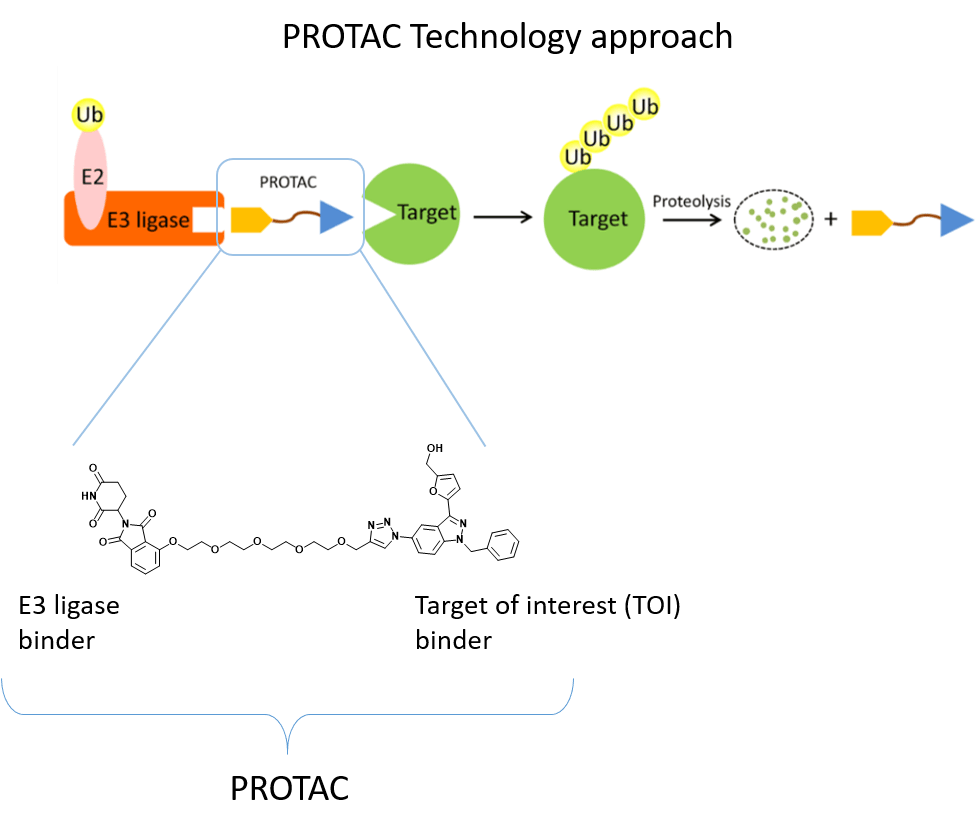
– The Ubiquitin-Proteasome System (UPS) consists of a multiple enzymatic step process with sequential action of ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3). The addition of polyubiquitin chains to a target protein serves as a recognition marker for its proteolytic degradation through the proteasome. Dysregulation of this system, such as the presence of non-functional ligases, may result in a disruption of this pathway and substrates overexpression.
Proteolysis Targeting Chimeras (PROTACs) represent a powerful tool to investigate the mechanisms behind UPS dysregulation in cancer. PROTACs are heterobifunctional molecules that specifically bind and bring into proximity a target protein and a given E3 ubiquitin ligase. Contrary to protein inhibition, PROTACs act as degraders by using the cell’s protein degradation pathway to remove specifically labeled proteins.
Evidence from the literature has shown that the lack of functional VHL E3 ubiquitin ligase in VHL disease causes the accumulation of the transcription factor HIF-2α, which promotes human cell renal cell carcinoma (ccRCC). For this reason, our project aims at developing PROTACs targeting HIF-2α to restore the protein degradation pathway and to counteract tumor growth.
Hit-to-lead cancer immunotherapy drug discovery of adenosine 2A receptor inhibiting small molecules
David Pejoski 1, Aurélie Gouiller 1, Margot Boujut 1, Camille Suess 1 and Leonardo Scapozza 1
1 School of Pharmaceutical Sciences, University of Geneva, Rue Michel-Servet 1, 1206 Genève
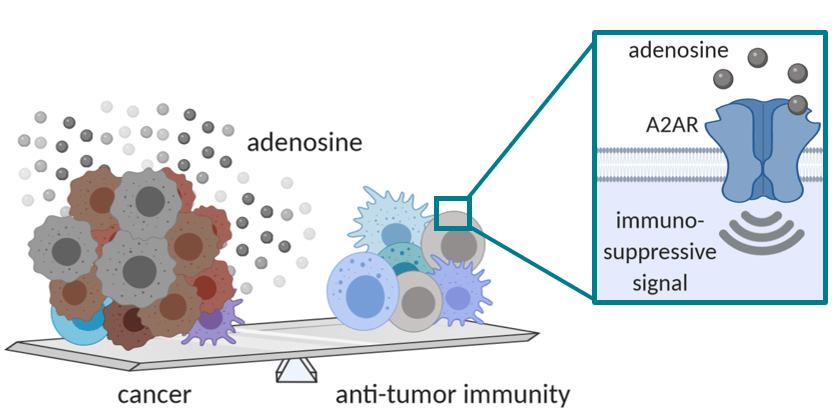
– Immune system-targeting therapies such as immune checkpoint inhibitors (ICI) show impressive clinical benefits in nearly 30% of cancer patients. To improve this response rate, new therapies aim to overcome the immunosuppressive tumor microenvironment. For example, high adenosine concentrations in tumors are known to deactivate tumor-targeting immune cells that express the adenosine 2A receptor (A2AR). Therefore, ideal A2AR blocking drugs should show activity regardless of high extracellular adenosine levels – a key feature of drugs classified as negative allosteric modulators (NAMs).
A team of researchers led by Dr. David Pejoski in the lab of Prof. Scapozza, are applying specialized (allosteric) drug discovery know-how to identify small molecules that block A2AR. The NAM drugs discovered thus far can block the effects of A2AR in vitro, with ongoing testing during an Innosuisse-funded project starting in December 2020 to improve the molecular structure (medicinal chemistry) of the NAMs. This will be followed by testing in biologically-relevant models to allow selection of ‘lead’ candidates for extensive toxicity testing and then human clinical trials. A small molecule A2AR NAM would be highly suited to treating a wide range of different cancers, especially if administered in combination with traditional and emerging therapies, including ICIs, CAR-T cells, cancer vaccines, as well as in a personalized medicine approach for patients with adenosine-rich tumors.
Synthesis and biological evaluation of PROTACs and destabilizers with putative anti-inflammatory activity
Sara Pannilunghi 1, Sébastien Tardy 1, Diana de Britto Salgado 1, Aurélie Gouiller 1, Laurent Galibert 2, Thibaut De Smedt 2 and Leonardo Scapozza 1*
1 School of Pharmaceutical Sciences, University of Geneva, Rue Michel-Servet 1, 1206 Genève
2 PATHIM, University of Geneva
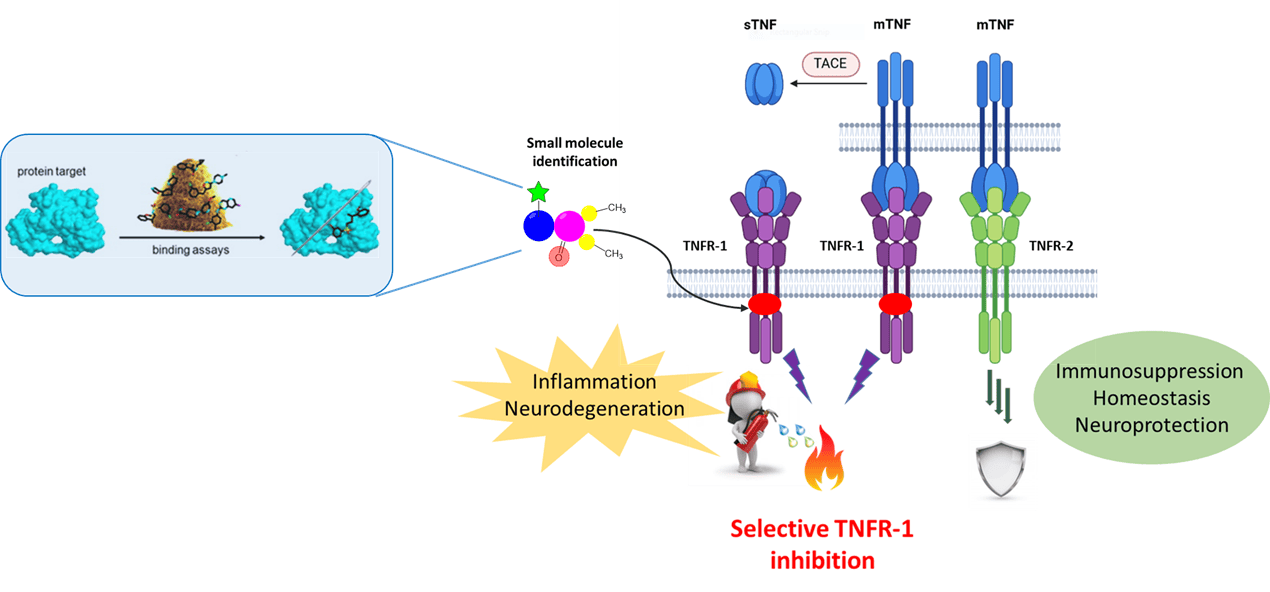
– Our research focuses on the identification of a selective way to deactivate the Tumor necrosis factor receptor 1 (TNFR1) in order to achieve a selective degradation of the receptor while preserving the protective effect of related isoform (TNFR2). Selective TNFR1 elimination would prevent the activation of the pro-inflammatory mediated cascade, responsible for the pathological consequences related to chronic inflammation and autoimmune disorders.
In order to reach this objective, two different strategies are adopted:
- The “small molecule” approach: Through a series of binding affinity and cellular assays, using readouts such as flow cytometry, BRET and ELISA for downstream signaling pathway monitoring, we have identified small scout molecules able to bind the region of interest (RoI) and causîng the elimination of TNFR1 at the cell surface in a dose-dependent manner. The first milestone is represented by target validation with the use of small scout molecules allowing the investigation of the RoI and the definition of the epitope to be targeted.
- The “proteolysis targeting chimeras (PROTACs)” approach: Selection of the most promising molecules binding to the RoI as putative target of interest (TOI) binders for PROTAC conception. Assessment of their cellular permeability and evaluation of their mechanism of action measured by TNFR1 degradation via western blot.
Scaffolds for Alternative Delivery (SADEL) for IBD and oncofitin’s platform

Nanofitin® scaffold. Residues (red) are randomly mutated to get the binding to the target of interest
Magali Zeisser-Labouèbe1 1, Aurélie Gouiller 1, Olivier Petermann 1 and Leonardo Scapozza 1
1 School of Pharmaceutical Sciences, University of Geneva, Rue Michel-Servet 1, 1206 Genève
NanoFitins® (NF) are small single-chain affinity proteins derived from the hyperstable DNA-binding protein Sac7d (7 kDa, 66 amino acids) of Sulfolobus acidocaldarius (see figure). High-affinity nanofitins® can be easily engineered as API themselves or targeting agents by ribosome-display over a wide range of targets (cell surface proteins, enzymes, cytokines etc.) by the full randomization of 10 to 14 amino acid residues localized in the DNA-binding site of Sac7d. In collaboration with Affilogic, Nanofitins® are explored in different projects involving challenging nonsystemic administration of biologics; such as in the Eurostars G2B project (Gut-to-Blood) or in the European SADEL (Scaffold for Alternative Delivery) project. The latter investigates orally deliverable anti-TNFα Nanofitins® as biotherapeutics for Inflammatory Bowel Diseases (IBD). The high physico-chemical and enzymatic stability provided by the Nanofitin® scaffold allows conserving their integrity in the harsh conditions of the GI tract (Figure 2) and thus NanoFitins® could overcome the challenge of oral administration of biotherapeutics. NanoFitins® can also be formatted as vectors for targeted drug delivery or crossing barriers for different applications. For example, in the Eurostars ODC project, our consortium are developing OncoFitin-Drug conjugates (ODC) for delivering cytotoxic payloads right into cancer cells by targeting receptors overexpressed in tumor cells.
Computer-assisted drug discovery for new cancer drugs
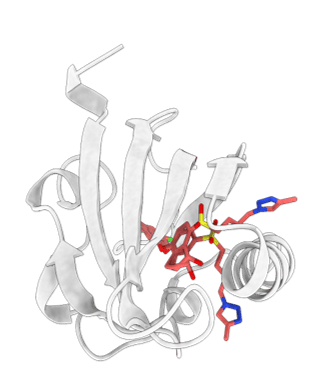
Margaux Héritier 1, Francesco L. Gervasio 1and Leonardo Scapozza 1
1 School of Pharmaceutical Sciences, University of Geneva, Rue Michel-Servet 1, 1206 Genève
The Drug discovery process usually involves many rounds of synthesis and in vitro testing. To complement and guide synthesis, we use modelling software to visualize the most important interactions between ligands and the targets. We also use molecular dynamic simulations, which acts like a molecular magnifying glass to get insight into the target dynamics at the atomic level. Computational tools helped design A2AR negative allosteric modulators, PROTACs against HIF2a, and helped understand the activation mechanism of TNFR1-DD.