Rare diseases
Targeting RNA stem-loop structure regulating the alternative splicing of RNAs linked to rare diseases e.g SMN2 (SMA) or LMNA (progeria)
Ece Sahi Ilhan 1, Olivier Petermann 1, Margaux Héritier 1and Leonardo Scapozza 1
1 School of Pharmaceutical Sciences, University of Geneva, Rue Michel-Servet 1, 1206 Genève
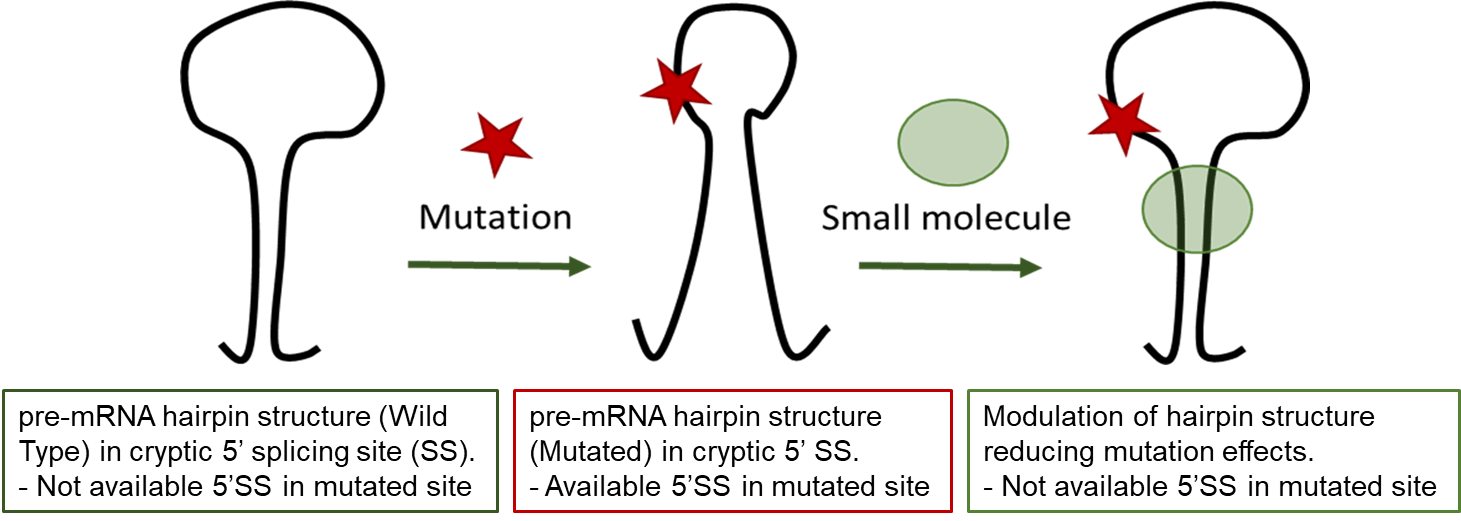
RNA splicing is a process where a recently made precursor mRNA (pre-mRNA) is transformed into a mature mRNA. Alternative splicing is a regulated process during which particular exons may be included within or excluded from the final mRNA. Alternative splicing is the main mechanism by which eukaryotes provide protein diversity. Aberrant splicing isoforms can also cause disease, such as Hutchinson-Gilford progeria syndrome due to a mutation in LMNA gene.
Hutchinson-Gilford progeria syndrome (HGPS) is a very rare, sporadic, autosomal dominant disorder resulting in early death. HGPS is caused by a point mutation in exon-11 of the LMNA gene. This mutation promotes recognition of a cryptic alternative splicing donor site in exon-11 and reduces the recognition of the authentic splicing site resulting in the production of Progerin, a truncated toxic protein leading to an aberrant nuclear morphology, genetic instability, and premature senescence. Progerin accumulates in the cell nuclei of HGPS patients and its accumulation is the biomarker of the disease. Symptomatic treatments of HGPS are available but no disease modifying treatment is existing.
In this work, to address the unmet medical need of HGPS, we aim at understanding the relationship between the conformational plasticity of pre-mRNA structure of exon-11 carrying the mutation around its cryptic splice site and the splicing behavior to ultimately validate this structural element as a target for small molecules that would abolish the production of progerin.
Pre-clinical evaluation or repurposed drugs and nutraceuticals in fatal inherited muscular disorders
Olivier Dorchies 1, Laurence Neff 1, Hesham Hamed 1, Ghali Guedira 1, Barbara Pinheiro 1, Oliver Petermann 1 and Leonardo Scapozza 1
1 School of Pharmaceutical Sciences, University of Geneva, Rue Michel-Servet 1, 1206 Genève
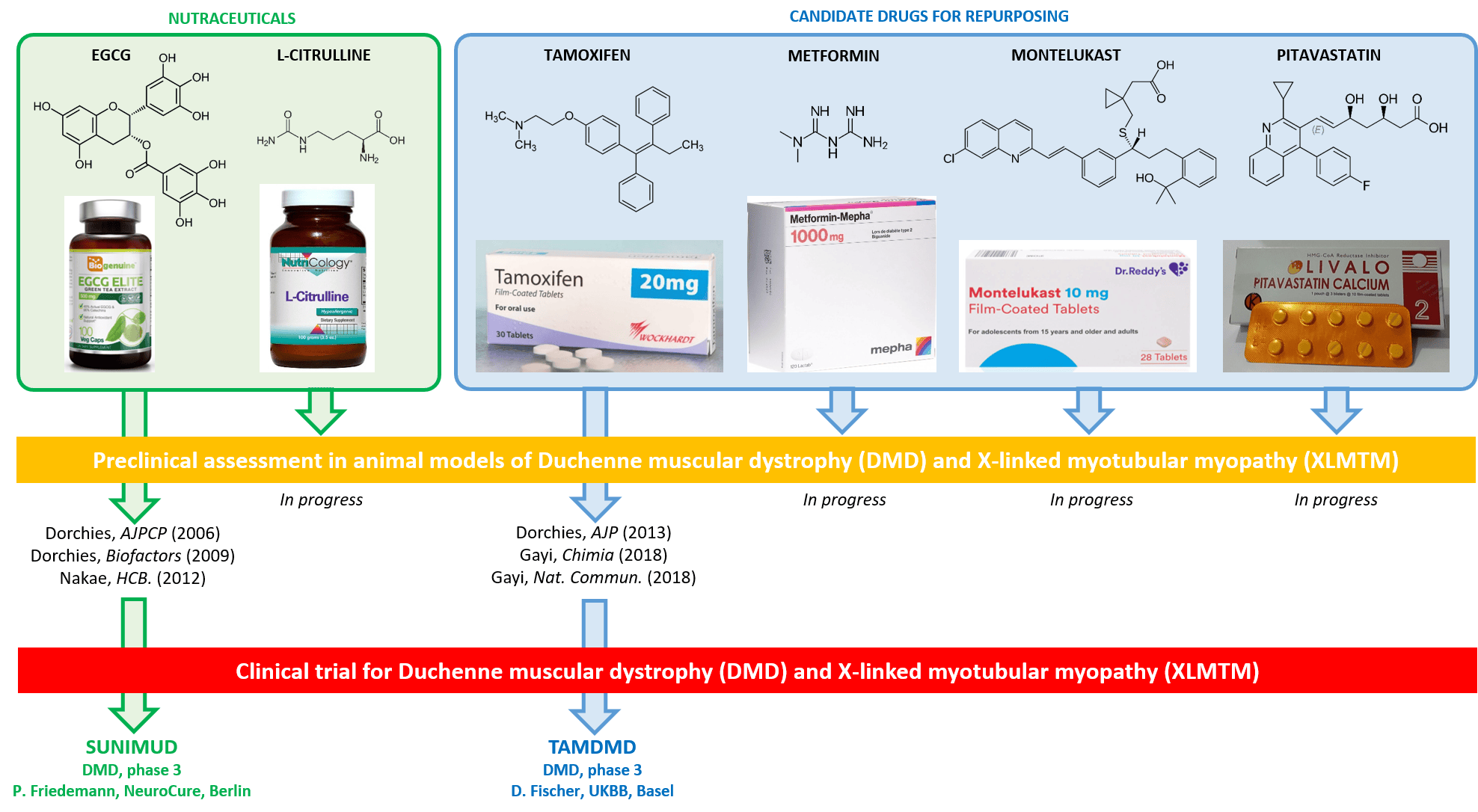
Duchenne muscular dystrophy (DMD) and X-linked myotubular myopathy (XLMTM) are rare X-linked diseases causing premature death of the affected males at around 25 and 2 years of age, respectively. In DMD, mutations in the gene encoding dystrophin cause muscle necrosis and improper regeneration, resulting in muscle wasting, paralysis and fatal cardiomyopathy. In XLMTM, the phosphatase MTM1 is absent, causing arrest of muscle maturation at the embryonic developmental stage, with severe hypotonia and respiratory distress from birth. Most innovative treatments for DMD and XLMTM, including AAV-mediated gene therapy are facing major cost, ethical and safety issues.
In order to address the major unmet therapeutic need for DMD and XLMTM we used a hypothesis-driven approach for carefully selecting a variety of nutraceuticals (citrulline, green tea polyphenols) and drugs (tamoxifen, metformin, pitavastatin, montelukast) suitable for repurposing. We are assessing these compounds, alone and in combination, in mdx/DMD and Mtm1KO murine models using a wide panel of complementary approaches comprising locomotor testing, analysis of gene and protein expression in tissues, histological investigation of muscle structure, quantification of biomarkers, and therapeutic drug monitoring. Using this strategy, we have previously identified EGCG and tamoxifen, and promoted phase 3 clinical trials in DMD boys.
Exploring the role of CEMIP axis in alport syndrome
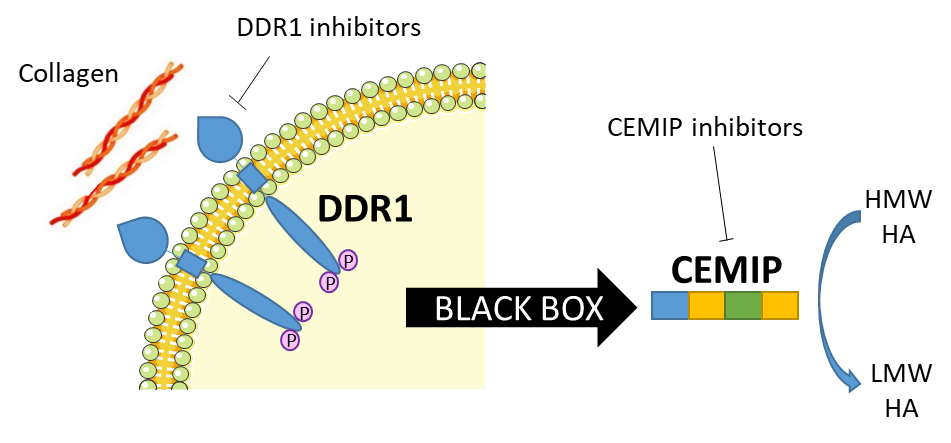
Intracellular pathway of the DDR1/CEMIP/HA axis
Sofia Spataro 1, Jacopo Sgrignani 2, Andrea Cavalli 2, Jonathan Sleeman 3, Anja Schmaus 3, Leonardo Scapozza 1 and Marco Prunotto 1
1 School of Pharmaceutical Sciences, University of Geneva, Rue Michel-Servet 1, 1206 Genève
2 Institute for Research in Biomedicine, Università della Svizzera Italiana
3 Universitätmedizin Manheim
Alport syndrome (AS) is a pediatric rare disease caused by mutations affecting type IV collagen, a major network-forming structural component of basement membranes found in the ear, eyes and kidneys. AS leads almost inevitably to loss of renal function or end-stage renal disease (ESRD), and until now the current standard of care aims to reduce or delay the progression of the disease, but lacks a specific therapy. Here we show a new approach in the study of the biology of this disease that includes the collagen receptor DDR1 and a newly identified hyaluronic acid (HA) degrading protein CEMIP (see figure) Could the DDR1/CEMIP/HA axis be a new way to address the medical need in Alport syndrome? To answer this question we want to better study and understand CEMIP by identifying amino acids essential for its activity. Exploring the role and the mode of action of CEMIP, for example by investigating its domains and its degradation product (HA fragments) composition and effect, will enlighten the cellular pathway that is compromised in Alport syndrome and will potentially enable the development of new inhibitors as treatments for this rare disease. A better understanding of this complex protein will empower research not only for Alport syndrome treatments but also in all the connected research areas where CEMIP has been found to have an essential function.