Research
Modelling of binding kinetics
We have been interested in a long time in the accurate prediction of binding kinetics and its optimization to increase the efficacy and decrease the toxicity of drugs. Our TS-PPTIS algorithm, which effectively combines Metadynamics, Path Collective Variables and transition path sampling, is able to predict the binding kinetics of ligands to their targets even in difficult cases, e.g. when multiple metastable states and conformational-selection or induced fit effects play a role. We have validated this approach in Src kinase using NMR and SPR, showing that the binding is a combination of induced-fit and conformational selection mechanisms (E. Pucheta-Martínez et al. Sci Rep 2016). Now we are applying this approach to predict the binding kinetics of various ligands to GPCRs and, in collaboration with Heptares, will use this knowledge to fine tune the on and off rates to increase their efficacy.
Obtaining atomistic insight that is difficult to obtain using conventional physical methods in order to optimise potential therapies
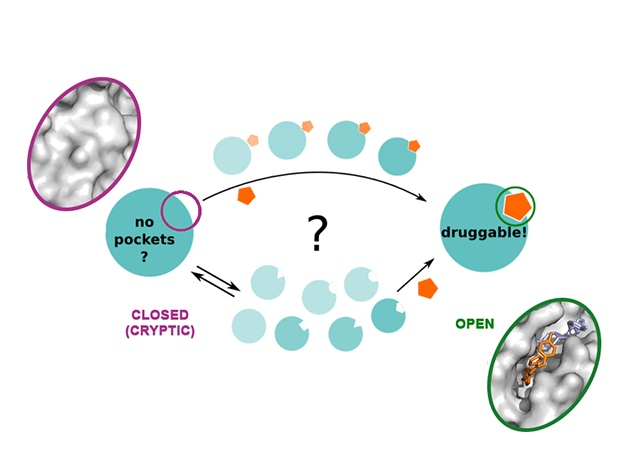
The algorithms we developed can be effectively used to predict the large-scale dynamics of proteins and other biomolecules, including druggable metastable states that might be difficult to capture by X-ray crystallography and other high-resolution experiments. Recently we developed SWISH, a new algorithm to understand the dynamics of cryptic pocket opening in proteins (V. Oleinikovas et al. JACS (2016), and F. Comitani et al. JCTC (2018)). Cryptic or hidden pockets are cavities that are not visible in the crystal structure of apo-proteins as they only open when a specific ligand bind. Thus, they are intrinsically difficult to find by standard experimental and computational approaches but offer outstanding opportunities to target proteins deemed ‘undruggable’ by classic substrate-competitive inhibitors. Our research, in collaboration with UCB, addressed the knowledge gaps in the dynamics of cryptic pocket opening and allowed the development of an efficient computational platform to systematically detect druggable cryptic pockets in targets of biopharmaceutical interest. The paper has attracted a lot of interest, including blog coverage from practical fragments, a blog on fragment-drug design and was a starting point for an EPSRC-funded proposal (£521,367) Efficient modelling and validation of cryptic protein binding sites for drug discovery.
Modelling of membrane permeation to optimise pharmacokinetics
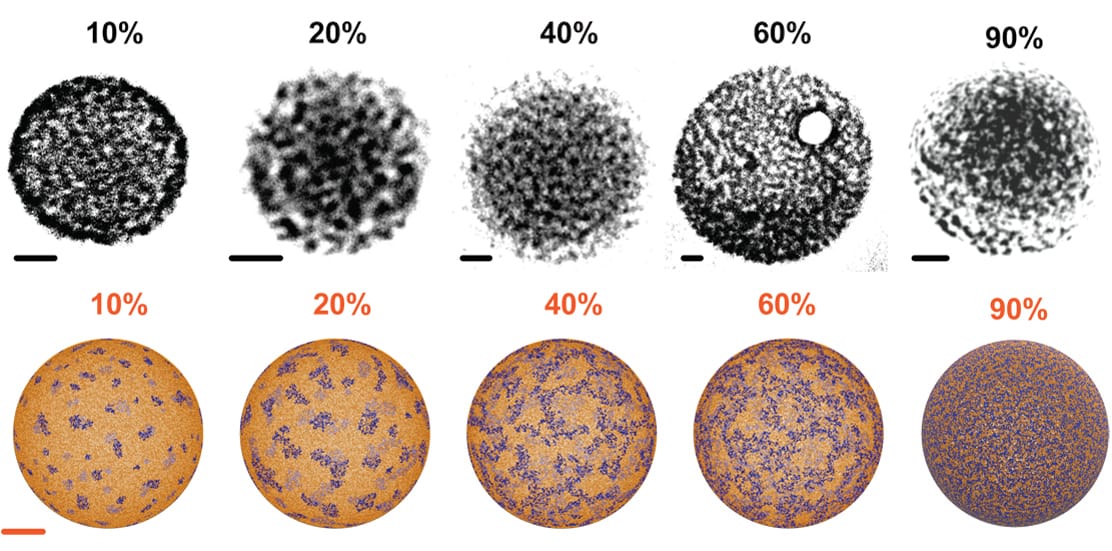
We co-developed with the group of Prof. Battaglia synthetic polymersomes with virus-like patterns that increase their membrane permeation potential and used a multi-scale simulation approach to engineer the interfacial tension within the polymersome surface (L. Ruiz-Perez et al. Science Advances 2016).
Provide a better understanding of the relationship between the amino acid sequence and function of proteins
Our group developed the first computational method based on evolutionary principles to predict protein dynamics. The approach combines an accurate contact prediction algorithm (from primary sequence alignments) and a protein coarse-grained model to explore conformational landscapes congruent with coevolution. We showed that both structural and dynamical properties can be already recovered using evolutionary information only. (L. Sutto et al. Proc Natl Acad Sci USA 2015).
Modelling the regulation of therapeutic proteins by allosteric mechanisms and post-translational modification
Our group studies Allostery and its therapeutic application. In 2016, we co-authored with R. Nussinov and others an influential review on the role of protein loops and linkers in conformational dynamics and allostery (E. Papaleo et al. Chem Rev (2016)). Recent highlights of our research in the field were:
- the clarification of the mode of action of the first allosteric inhibitor (SSR) of the Fibroblast Growth Factor Receptor. The enhanced-sampling simulations and NMR experiments have shown that SSR induces a previously unknown conformational change in the extracellular portion of FGFR opening a cryptic pocket. The accurate structural information obtained from the simulations was used by Sanofi and EVOTEC to design SSR derivatives now being developed as anti-angiogenic and anti-cancer agents (F. Bono et at. Cancer Cell (2013), and C. Herbert et al. Cancer Cell (2016)). With EVOTEC we are now studying how SSR modulates the protein-membrane interactions (to be submitted to a high impact journal).
- by using enhanced-sampling simulations, NMR and biophysical experiments we were able to reconcile different views on the mode of action of cancer-causing and drug-resistance-causing mutations in pharmaceutically relevant kinases.
- by combining structural experiments (X-ray crystallography) and simulations, we clarified the mode of action of allosteric inhibitors of choline kinase PDK1 and activating allosteric ligands of the PDK1 protein kinase (Jörg O.Schulze et al. Cell Chem Biol (2016)).
- combining simulations and NMR experiments we have clarified the effects of phosphorylation and the nature of an allosteric cross-talk between the phosphorylation and the active site in Src kinase and p38α MAP kinase (A. Kuzmanic et al. eLife (2017)). Currently, we are studying the effect of phosphorylation on the structural and dynamical properties of Huntingtin.
An unexpected role for the enzyme Glutamine Synthetase
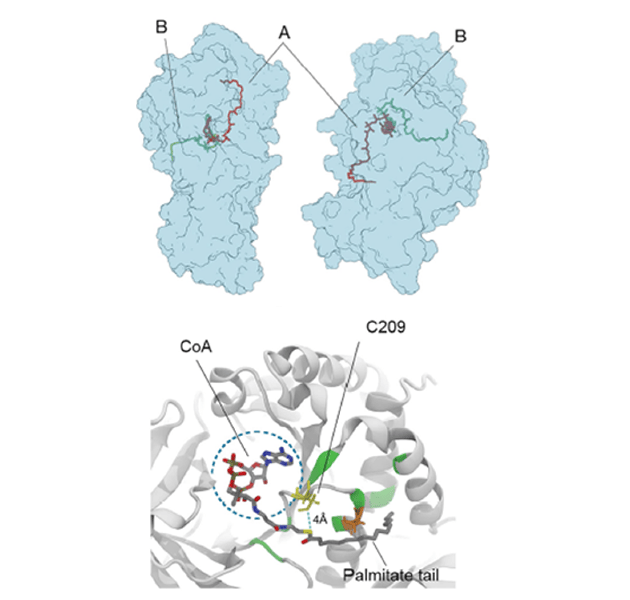
Our group contributed to discover and clarify an unexpected, yet fundamental, role of the enzyme glutamine synthetase. Glutamine synthetase (GS) is an enzyme that converts glutamate and ammonia to glutamine. GS is expressed in endothelial cells, fundamentally regulating vascular development. However, a group of scientists led by Prof. Peter Carmeliet at VIB in Belgium together with Prof. Francesco Luigi Gervasio at UCL found that it surprisingly shows little glutamine synthesizing activity in these cells. Instead, GS localizes in membranes due to an expected (and so-far, unknown) auto-palmitoylation activity. This moonlighting activity turns out to be fundamental for vessel sprouting. However, it was unclear how it happens and which residues are involved. Using molecular dynamics simulations, we were able to solve the mystery, showing how palmitoyl-COA binds to the active site and reacts with cysteine 209 to form a covalent bond. Site-directed mutagenesis of Cys209 later confirmed the computational prediction and validated the proposed mode of action. This new finding, published by the prestigious journal Nature (G. Eelen et al. Nature (2018)), has important implications for anti-cancer drug discovery, as it might lead to new drugs blocking the vascular development in solid cancers.